治験モニターは危険・怖い?知っておくべきリスクと安全への取り組み
治験は、高額な負担軽減費や自由時間の長さが魅力的だけれど「ちょっと危険なやばいアルバイト」として認識されていることがあります。
一般的なアルバイトよりも高額な負担軽減費(協力費)を得られることから興味はあるけれど。。。開発中の薬の安全性や副作用に不安を感じてしまい、ちょっと「怖い」・「不安」という感覚が払拭できずに応募や登録をためらっている人も多いのではないでしょうか?
今回は治験モニターの一般的なアルバイトとの違いや安全への取り組みについて紹介します。治験とは何かを正しく理解して参加への判断基準にしてみてください。
目次
1.臨床試験と治験の違いとは
病気で困っている患者さんがその誕生を心待ちにしている新薬は、長い年月をかけて開発されています。その開発の最終段階が「治験」です。
まず、「臨床試験」とは、新しい薬や治療法、医療機器などが人に対して安全で有効であるかを確認するために行われる研究全般を指します。病気の予防、診断、治療の改善を目的として医療機関や研究機関で実施されます。 この臨床試験の中に、「治験」が含まれます。治験とは、厚生労働省から新しい薬として承認を得ることを目的として行われる臨床試験のことです。つまり臨床試験はより広い概念であり、治験はその一部であると理解すると良いでしょう。治験は国が定めた厳格なルール(GCP:医薬品の臨床試験の実施の基準)に従って実施され、参加者の安全が最優先されます。治験以外の臨床試験も、同様に倫理的な配慮と科学的妥当性に基づいて実施されます。
臨床試験は新しい医療の発展に不可欠であり、参加することで社会貢献(医療の発展に貢献)ができる貴重な機会です。また、ご自身の健康状態を医者にチェックしてもらえたり、負担軽減費(協力費)を受け取ることができます。
1-1.治験が重要な理由
日本国内でお薬を販売するためには、厚生労働省から許認可を受けなくてはなりません。そのため、製薬会社は医療機関と協力して、厚生労働省の定めたルール(GCP:医薬品の臨床試験の実施の基準)に基づき、薬の認可を目的とした臨床試験を行います。
治験(ちけん)とは、新しい「くすり」や「医療機器」が国の承認を得て世の中に出るために、人での効果(有効性)や安全性(副作用など)を確認する臨床試験(研究のための治療)のことです。
私たちが普段、病院や薬局でもらっている医薬品は、すべてこの治験を経て安全性と有効性が証明されたものです。
また、副作用やリスクに関する情報を収集し、データ化することも治験の目的の1つです。治験の実施によって得られたデータは、実際に薬を処方するときの確かな指針にもなります。
将来、より良いお薬が使用できるようになるためには、医師や研究者はもちろん、治験に参加いただく治験ボランティア皆さんの協力が欠かせないのです。
1-2.新薬が開発の流れと治験モニターの役割
新薬の開発は、一般的に次のような手順で進めていきます。
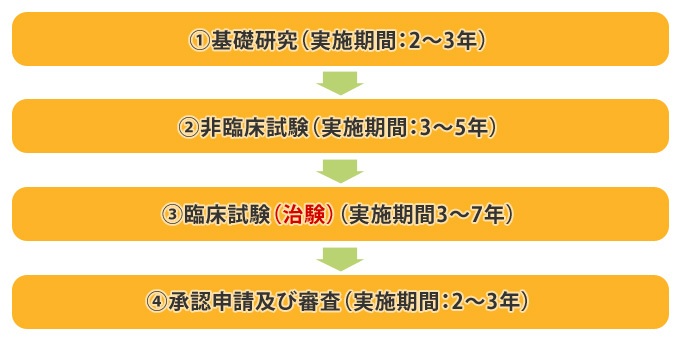
基礎研究では物質や新たな成分の組み合わせを研究して、薬の候補を試作します。基礎研究を経てその薬の候補の有効性や安全性が確認されると次に非臨床試験(動物での試験)を実施し、効果や安全性を徹底的に検討します。
しかし、非臨床試験だけではヒトへの効果がわかりません。そのため、ヒトに安全である容量を設定できた段階で、臨床試験(ヒト試験)に移ります。動物実験の結果から、ヒトに初めて投与する安全な用量(推定開始用量)を決定し、ごく少量から徐々に用量を増やしていきます。
この臨床試験に被験者として参加するのが治験モニターなのです。(これらの被験者は医学・創薬の業界では治験モニター・医学ボランティア・創薬ボランティアなどと呼ばれています。一般には治験アルバイトとか治験バイトとして馴染みがあるかもしれませんが、アルバイトとは異なります。)
お薬はヒトが使うものなので必ず臨床試験(ヒト試験)によってその有効性や安全性を証明しなければならないのです。
2.治験モニターが知っておくべきリスクについて
治験にリスクがまったくないと言い切ってしまう事は不誠実です。
しかし、日本で行われている治験の多くはジェネリック品(既に10年以上販売されているお薬と同成分の薬)や既に海外では利用されているものを日本国内で販売する承認を得る為に行われる治験(First In Japanese:FIJ)試験)が多く、実は多くの人がイメージしている治験=人類で初投与という治験(First in human:FIH)の実施数はあまり多くあまりません。
日本国内で行われている治験をしっかりと理解することで、治験モニターへの漠然とした不安は払拭されるはずです。
2-1.治験における3つの段階
治験は次の3種類に分類され、段階的に実施されていきます。
| 段階 | 対象となる被験者 | 主な内容 |
|---|---|---|
| フェーズ1 (第1相試験) |
健康な成人 | 薬の吸収と代謝や安全性を評価する |
| フェーズ2 (第2相試験) |
対象となる疾患を持った患者(少数) | 治療薬としての有効な投与量、及び投与方法を検討する |
| フェーズ2 (第3相試験) |
対象となる疾患を持った患者(多数) | 第2相試験決められた容量・使用法で多数の患者を対象に有効性と安全性の評価を行う |
このフェーズとは治験の安全性のレベルを表すものではなく、臨床試験が確認しようとする内容によって分けられています。
健康な人が事前健康診断を受診し、参加いただいている多く治験はフェーズ1段階の治験になります。インターネットのサイトなどで広く募集されている治験モニター(一般に治験アルバイト・治験バイトなどと呼ばれている)はこのフェーズ1治験が該当します。
フェーズ1治験の安全性がどうしても心配な方は、同じフェーズ1治験の中でも既に発売されているお薬の治験「ジェネリック薬の治験」に参加することをオススメします。
2-2.「有害事象」と「副作用」

治験で起こり得るリスクとして挙げられるのが、有害事象と副作用です。
有害事象(Adverse Event: AE)とは、治験薬(または医薬品)を投与された被験者に生じた「ありとあらゆる好ましくない、または意図しない徴候、症状、あるいは病気のこと」です。
※治験薬との因果関係の有無は問わない
治験で入院中に風邪をひいて熱が出た場合や通院の途中に発生したケガ、1期目と2期目の間で帰宅中に発生した食あたりなども有害事象に該当します。治験や臨床試験の現場では、まず「有害事象」としてすべてのトラブルを記録し、その中から専門家が「薬が原因である(因果関係が否定できない)」と判断したものが「副作用」として登録されます。
副作用(Adverse Drug Reaction: ADR)とは、治験で生じた有害事象のうち、「治験薬との因果関係が否定できない(少なくとも合理的な可能性が否定できない)もの」を指します。治験参加時に想定されている副作用に関しては同意説明会(インフォームド・コンセント)によって参加者へ事前に説明されます。
なぜ治験ではここまで厳密に有害事象と副作用を分けるのでしょうか?
理由は、「見落としを防ぐため」と「国際的な安全性のシグナルを検知するため」です。もし、最初から「これは薬のせいじゃないだろう」と医師の主観で勝手に有害事象を間引いて記録してしまったりすると、「実は世界中でも同じ副作用が発生していた」という重大なリスク(シグナル)を見落とすことになってしまいます。
なので治験では
1:まずは「有害事象」として、起きたことを漏れなくすべて集める(網羅性)。
2:その中から、薬との関係が疑われるものを「副作用」として分析する(因果関係)。
3:さらに命に関わるようなものを「重篤な有害事象/副作用(SAE/SADR)」として国へ緊急報告する。
という3ステップの安全網を敷いています。この安全網を敷くためにGCP省令やICHガイドラインでは「有害事象と副作用」という言葉の定義と境界線を厳格に管理しているのです。
厚生労働省は平成24年に「医薬品の臨床試験の実施の基準に関する省令」のガイダンスについてを改正しました。このガイダンスでは副作用の判断するための参考事例として、投薬中止後の症状の消失や、投薬再開後の症状再発などを挙げています。以降、副作用の判断基準はこのガイダンスの参考事例に照らし合わせて行うこととなっています。
3.治験での「安全」に対する取り組み・補償
予期せぬ副作用が生じた場合の影響を最小限に抑えるために、治験では医療機関や製薬会社、そして国が連携して治験モニターの安全に対する取り組みを行っています。
有害事象が起こった際も被験者の安全を最優先に緊急処置・報告の手順が提唱されており、医療機関は国の安全基準やルールを守らなければなりません。
万が一重篤な副作用で健康に被害が生じた場合は、しっかりとした治療と金銭的な補償が行われます。
3-1.治験を安全に行うための取り組み・制度

《ヘルシンキ宣言》
現代の医学・薬学では最終的にヒトを対象とした試験によらなければ確認できない事柄がたくさんあります。つまり治験や臨床試験を行うことは医学・薬学の発展の為にまだまだ必要です。その為、世界医師会は主にこの研究に携わる医師に対し、過去の人体実験への反省を鑑み、ヒトを対象とする生物医学的研究について倫理的基盤を定めました。
これをヘルシンキ宣言と呼んでいます。この宣言は1964年に制定されて以来、定期的に見直し検討されています。
特に重要なこととして、・科学的・倫理的に適正な試験実施計画・治験審査委員会による科学的・倫理的な適正さを審査・参加者へ十分な説明と自由意思による同意等が謳われています。
《GCP(good crinical practice)医薬品の臨床試験の実施の基準》
治験は、ルールに基づいて事前に厚生労働省へ実施計画を届け出た上で遵守しなければなりません。
(このルールのことを《GCP(good crinical practice)医薬品の臨床試験の実施の基準》と言います。)
そのため、専門的な知識を持った製薬会社の担当者が、医療機関へ何度も足を運びます。担当者の役目は、治験におけるデータの科学的な正当性や信頼性の確認、そして被験者の人権や安全の確保を通じて、治験が適切に実施されるよう適宜調整することです。
《治験審査委員会(IRB)》
一方、治験の実施計画を審査しているのが、治験審査委員会です。実施計画を承認したあとも、治験審査委員会は年に1回以上、治験が適切に行われているかどうかを審査しています。
もし、治験の実施中に後遺症が残るなどをはじめとする重篤な副作用が起こった場合、医療機関はただちに治験審査委員会と製薬会社へ報告をしなければなりません。報告を受けた委員会は治験の継続可否について審査を行います。また、製薬会社は定められた期間内に、さらに国へ報告をしなければなりません。
このように、治験では医療機関、製薬会社、治験審査委員会、そして国といった複数の立場の関係者が絶えず連携することで、実施計画の管理や安全性の確保を行っています。
《インフォームド・コンセント》
もちろん治験に参加を希望する方には参加前に詳しく当該治験の内容について説明があります。その上で治験への参加は希望者本人が決めることになります。強制などはございませんので、一度同意(参加の意思表示)をした場合でも、いつでも取りやめることができます。
治験の実施中に認められた未知の副作用に関する情報は、参加中の他の被験者にも説明されます。併せて、治験の参加を継続するかどうかの意思を確認されるため、不安や怖さを感じた場合は止めることも可能です。怖いと思ったらその場でスタッフに伝えましょう。なお、一部の例外を除いて、基本的に治験はいつでも止めることができます。
3-2.治験によるリスクに対する補償
厚生労働省が定めた指針《GCP》に基づき、治験の依頼者は健康被害を補償するための要件や手続きを定めなくてはなりません。ここでいう健康被害とは、主に副作用によるものを指します。
治験の補償には、次のようなものがあります。
| 補償の種類 | 主な支給内容 |
|---|---|
| 医療費 | ・健康被害に対する治療費のうち、健康保険などからの給付を除いた自己負担額分 |
| 医療手当 | ・通院にかかる交通費 ・入院に伴う諸雑費 |
| その他補償費 | ・一定以上の障害を負った場合に支給する「障害補償金」 ・死亡した場合に遺族へ支払う「遺族補償金」 ・療養により休業し仕事ができない場合に支給する「休業補償金」 |
もちろん、副作用の確認は厳格に行われます。
しかし、副作用とは「新薬との因果関係が明らかなもの」に限りません。そのため、治験との関係性が完全には否定できないものに対しても、幅広く補償される可能性があるといえるでしょう。
また、因果関係の証明については被験者の負担にならないよう、治験依頼者が行うことになっています。因果関係を恣意的に判断することは禁止されているため、治験依頼者は誠実に検査を行い、客観的なデータを示すことが義務づけられています。
まとめ
治験に用いられる新薬は、理論上は危険がないことをあらかじめ徹底的に確認しています。また、日本国内では未認可であるものの、海外では既に使用されている新薬も多いのです。
万が一のリスクに対しても、参加者への安全に対する取り組みや補償が厳格に定められています。このことを意識すれば、治験モニターへの参加が不用意に怖いと思うものではないことがわかります。
風邪薬を飲むと眠くなるように、人体への副作用を全く持たない薬は基本的に存在しません。そのため、副作用という言葉だけを必要以上に不安がることはないでしょう。紹介したリスクや安全への取り組みから治験が安全だと感じたならば、ぜひ治験モニターへ応募してみましょう。














